Help & Documentation
Learn how to use PFGPred for fusion gene prediction and training
Background of the Project
Traditional gene fusion detection tools often show high false-positive rates in plants due to algorithms designed for human cancer datasets with different genomic structures. PFGPred is a robust deep learning–based model tailored for fusion gene discovery in plants, trained on high-confidence datasets validated with RNA-Seq and Whole-Genome Sequencing (WGS) data.
What are Fusion Genes?
Fusion genes are hybrid genes formed by combining sequences from two or more distinct genes. In plants, they play a critical role in stress response, gene evolution, and the development of novel traits, making their systematic detection essential for understanding gene regulation, adaptation, and crop evolution.
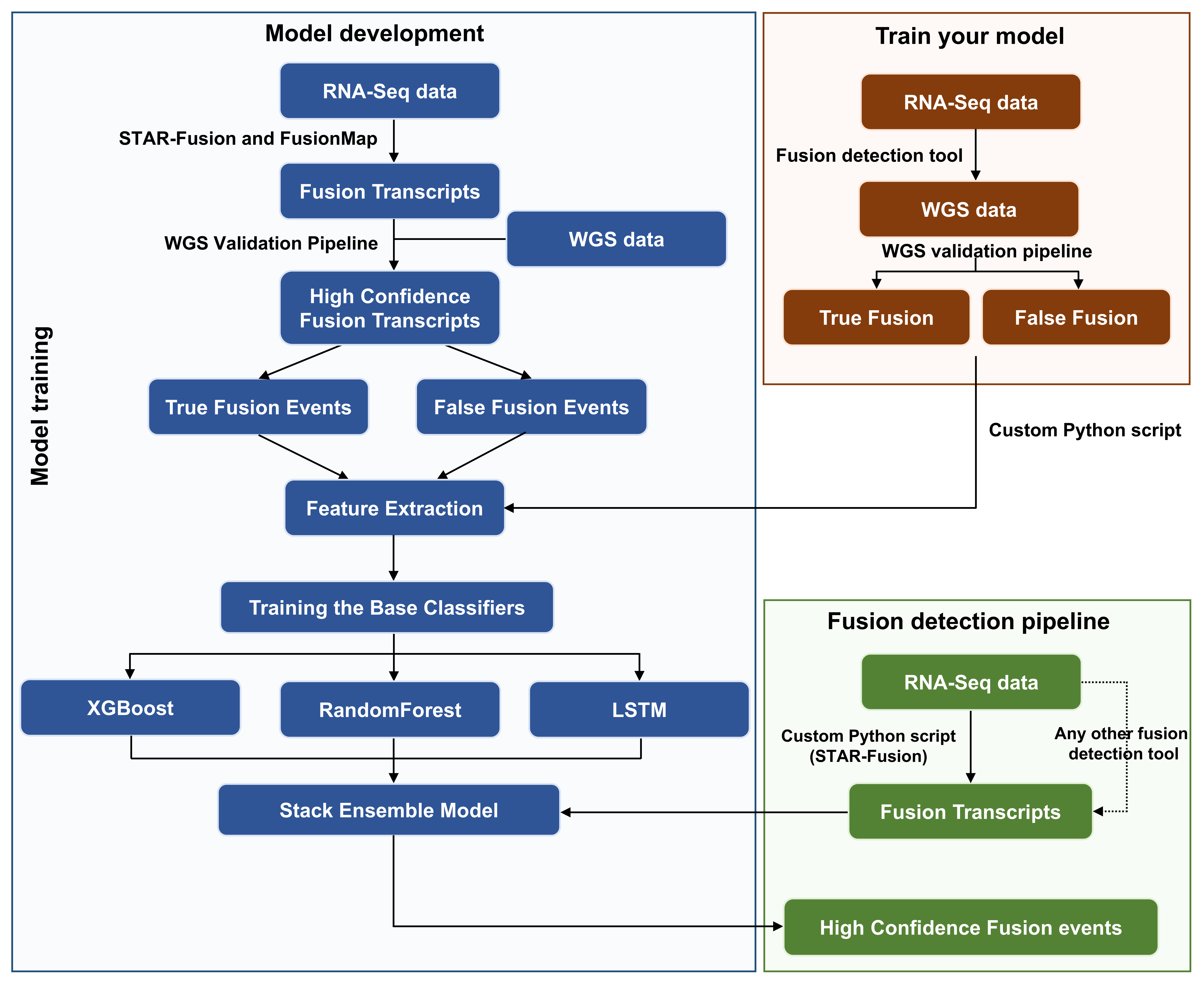
Overview of PFGPred pipeline and fusion gene detection workflow.
Frequently Asked Questions (FAQ)
Detailed answers and video guides for advanced usage.
1. How to use PFGPred?
PFGPred operates in two main steps:
- Fusion Detection and Feature Extraction: Use the custom Python script provided on GitHub to detect fusion transcripts from RNA-Seq data and extract the corresponding features. This step generates a feature table in CSV format.
- Prediction: Upload the generated feature table to the “Prediction” section of the PFGPred web server to obtain fusion gene predictions and confidence scores.
2. What type of input data is required for PFGPred?
PFGPred requires RNA-Seq data in FASTQ. Both single-end and paired-end reads are supported.
3. Can I use RNA-Seq data from any organism?
PFGPred is trained on Arabidopsis thaliana, Oryza sativa, Zea mays, and Triticum aestivum. Apart from these species, the model performance is also tested on Cicer arietinum, Glycine max, and Setaria italica. So the model showed decent performance for all the mentioned species.
4. Does PFGPred require WGS data for fusion validation?
No. Once trained, PFGPred uses only RNA-Seq data for fusion gene detection.
5. What should be the ideal threshold for high-confidence fusion gene prediction?
The default confidence threshold in PFGPred is 0.9, which generally provides a good balance between sensitivity and specificity. However, our evaluations across different species indicate that the optimal threshold may vary depending on the organism. Users are encouraged to adjust the threshold based on their desired stringency level.
6. How can I retrain the model for a new species?
Retraining PFGPred for a new species requires both RNA-Seq and whole-genome sequencing (WGS) data to ensure accurate model calibration. The retraining process involves the following steps:
- Fusion Detection and Feature Extraction: Use the custom Python script available on GitHub to detect fusion transcripts from RNA-Seq data and extract corresponding features. This step generates a feature table (CSV format) containing expression-, sequence-, and structure-based features for each candidate fusion.
- Validation of Fusion Events Using WGS: Validate the detected fusion events using the WGS validation pipeline (available on GitHub). This step helps distinguish true fusion events from false positives.
- Construction of Positive and Negative Datasets:
- Positive dataset: WGS-validated fusions
- Negative dataset: Non-WGS-validated fusions
- Model Training: Upload the positive and negative datasets to the “Prediction & Training” section of the PFGPred web server. The system will train a new model specifically optimized for the target species.
- Prediction for the Target Species: Once trained, the new model can be used to predict fusion genes using RNA-Seq data alone, without requiring additional WGS validation.
Data Format Requirements
* Your CSV file must include the following columns for processing.
Key Feature Columns:
- LeftBreakpoint
- RightBreakpoint
- FFPM
- Total_Count_(SC+RC)
- Splice_Site
- Total_Mapped_Reads
- Left_Exon
- Right_Exon
- 5_gene_start
- 5_gene_end
- 5_gene_length
- 3_gene_start
- 3_gene_end
- 3_gene_length
- alternate_junction_count
- exon_count5
- exon_count3
- LeftStrand_+
- LeftStrand__
- RightStrand__
- RightStrand_+
- Chromosome_Feature_Interchromosomal
- Chromosome_Feature_Intrachromosomal
- Same_Strand_No
- Same_Strand_Yes
- Reciprocal_Fusion_Yes
- Reciprocal_Fusion_No
- Splice_Pattern_InFrame
- Splice_Pattern_FrameShift
- Splice_Pattern_Unknown
- Splice_Pattern_Class_CanonicalPattern
- Splice_Pattern_Class_NonCanonicalPattern
- 5_loc_M
- 5_loc_S
- 5_loc_E
- 5_loc_O
- 3_loc_M
- 3_loc_S
- 3_loc_E
- 3_loc_O
- alternative_junction_Yes
- alternative_junction_No
* For training, include a target column (e.g., "label") with 1 for true fusions and 0 for false fusions.
Contact Support
For technical support or research inquiries, contact the SKLab at shailesh@nipgr.ac.in
support@PFGPred.example.com